
Research News: Aβ43 Generation in Familial Alzheimer’s Disease: New Insights into Disease Onset
January 07, 2022
Study reveals unique mechanisms of amyloid beta generation in familial forms of Alzheimer’s caused by mutations in presenilin 1
Alzheimer’s disease (AD) is caused by amyloid beta (Aβ) proteins, which are generated through the action of the enzyme γ-secretase. A new study has identified a novel “switch” for the production of Aβ43 in cases of familial AD caused by presenilin 1 mutations—a mechanism different from that observed in sporadic AD. Furthermore, the findings show that such Aβ43 generation is onset-dependent, indicating its potential in AD detection and treatment before disease onset.
Alzheimer’s disease (AD) is a progressive neurological disease that causes impairments in memory and cognition. AD is believed to result from the deposition of protein “plaques” consisting of amyloid beta (Aβ) proteins sequentially generated via the enzyme γ-secretase. Although most cases of AD are sporadic or late-onset, early-onset or familial AD (FAD) caused by mutations in genes encoding γ-secretase is also noted. Differences in the sequential processing of Aβ proteins between sporadic AD and FAD have so far remained unclear.
In a recent study published in Translational Psychiatry, researchers from Japan sought deeper insights into the molecular mechanisms of Aβ generation and γ-secretase activity in FAD. They systematically examined the levels of different Aβ proteins in patients with FAD caused by mutations in presenilin 1 (PS1), a gene encoding part of the γ-secretase complex. Dr. Nobuto Kakuda, who led this study, comments, “The onset of FAD occurs between the ages of 40 and 60 years due to different Aβ proteins. Although certain Aβ proteins are predominantly generated in FAD, the mechanisms underlying the alterations were unclear. Our study aimed to bridge these gaps.”
Given that Aβ proteins are sequentially generated in patients with AD, the researchers first examined the concentration of Aβ38, Aβ40, Aβ42, and Aβ42 in the cerebrospinal fluid (CSF) of patients with FAD and PS1 mutations. “In the brain, there are two pathways of Aβ production. First, the amyloid precursor protein is cleaved to generate Aβ49 and Aβ48. Aβ49 is further cleaved to Aβ46 and Aβ43 and then to Aβ40. In contrast, Aβ48 is cleaved to Aβ45 and Aβ42 and then to Aβ38. So, the levels of these proteins can help us understand the status of Aβ and AD progression,” explains Dr. Kakuda. Interestingly, the researchers found that in some patients with FAD, although low levels of Aβ38, Aβ40, and Aβ42 were detected in the CSF, the levels of Aβ43 appeared unchanged, indicating stable Aβ43 generation.
The researchers went on to validate these findings in cultured cells expressing a mutant form of PS1. Using in vitro γ-secretase assays, they confirmed that although Aβ38, Aβ40, and Aβ42 generation is lower in cells with PS1 mutations than in wild-type cells, Aβ43 generation is unaffected. Moreover, ratio between the level of Aβ43 and the level of the amyloid precursor protein intracellular domain (AICD) was elevated in these cells, further indicating increased Aβ43 generation. Together, these findings provided strong evidence supporting their initial hypothesis — the mechanism of Aβ processing appeared to be different in FAD compared to sporadic AD.
Next, the team used liquid chromatography–mass spectrometry to understand these altered mechanisms in more detail. Their findings revealed that in PS1 mutants, Aβ43 was not generated only from Aβ46, as in sporadic AD, but also from Aβ48. “We measured the release of various peptides generated during Aβ generation using mass spectrometry. In this way, we could find the source of the additional Aβ43 generated in cases of FAD,” says Dr. Kakuda.
Fortuitously, the researchers stumbled upon an even more interesting phenomenon. They found that the ratio of VIVIT, a peptide generated during Aβ48→Aβ43 cleavage, to AICD, a part of the amyloid precursor, changed with the age of FAD onset. A higher VIVIT/AICD ratio—which indicated a higher stage of disease progression—was correlated with an earlier age of onset. “The identification of this correlation marks a significant milestone in AD research. By measuring VIVIT levels, we could predict the age of onset in patients with a family history of FAD! That will allow us to perform preventive interventions even before disease onset,” quips Dr. Kakuda, excitedly.
Indeed, the findings by Dr. Kakuda and his team provide new insights into AD pathogenesis. A cure based on these newly identified mechanisms could be just around the corner!
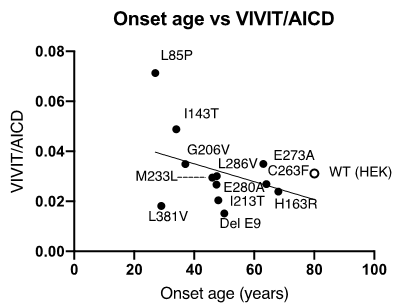
Correlation between the age of FAD onset and the VIVIT/AICD ratio.
In this study, γ-secretase was extracted from wild type (WT) cells and cells carrying FAD-related genetic mutations (mutations in presenilin 1: L85P, I143T, H163R, L166P, G206V, I213T, M233T, C263F, E273A, E280A, L286V, G384A, and delta E9). The enzyme was mixed with its substrate in vitro to generate Aβ, the amyloid intracellular domain (AICD), and other peptides. Given the varying levels of Aβ in the samples, the data were normalized to AICD levels. The ratio of VIVIT (peptide released during the enzymatic cleavage of Aβ48 to Aβ43) to AICD was correlated with the age of onset.
The open circle indicates WT enzyme, and the closed circles indicate FAD mutant enzymes. All experiments were repeated in triplicate.
Image courtesy: Nobuto Kakuda from Doshisha University
Reference
| Title of original paper | Switched Aβ43 generation in familial Alzheimer’s disease with presenilin 1 mutation |
| Journal | Translational Psychiatry |
| DOI | 10.1038/s41398-021-01684-1 |
| Latest Article Publication Date | 3rd November 2021 |
| Method of Research | Experimental study |
| Subject of Research | Cells |
| Conflict of Interest Statement | The authors declare no competing interests. |
Funding information
This study was partially supported by the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development (AMED) under Grant Numbers JP20dm0107128 (to N.K.), JP20dm0207073 (to Takeshi Ikeuchi. Niigata Univ.), and JP21dk0207047 (to T.I.).
Profile

Dr. Nobuto Kakuda has been an Assistant Professor at the Department of Medical Life Systems, Doshisha University, since 2015. He completed his PhD from the University of Tokyo and received postdoctoral training at Katholieke Universiteit Leuven, Belgium. His research interests include Alzheimer’s disease and pathological processes involving amyloid beta and gamma-secretase. He has published several papers in journals of high international repute, including the Journal of Biological Chemistry, EMBO Molecular Medicine, and The American Journal of Pathology.
Nobuto Kakuda
Assistant Professor, Faculty of Life and Medical Sciences, Department of Medical Life Systems
Media contact
Organization for Research Initiatives & Development
Doshisha University
Kyotanabe, Kyoto 610-0394, JAPAN
CONTACT US